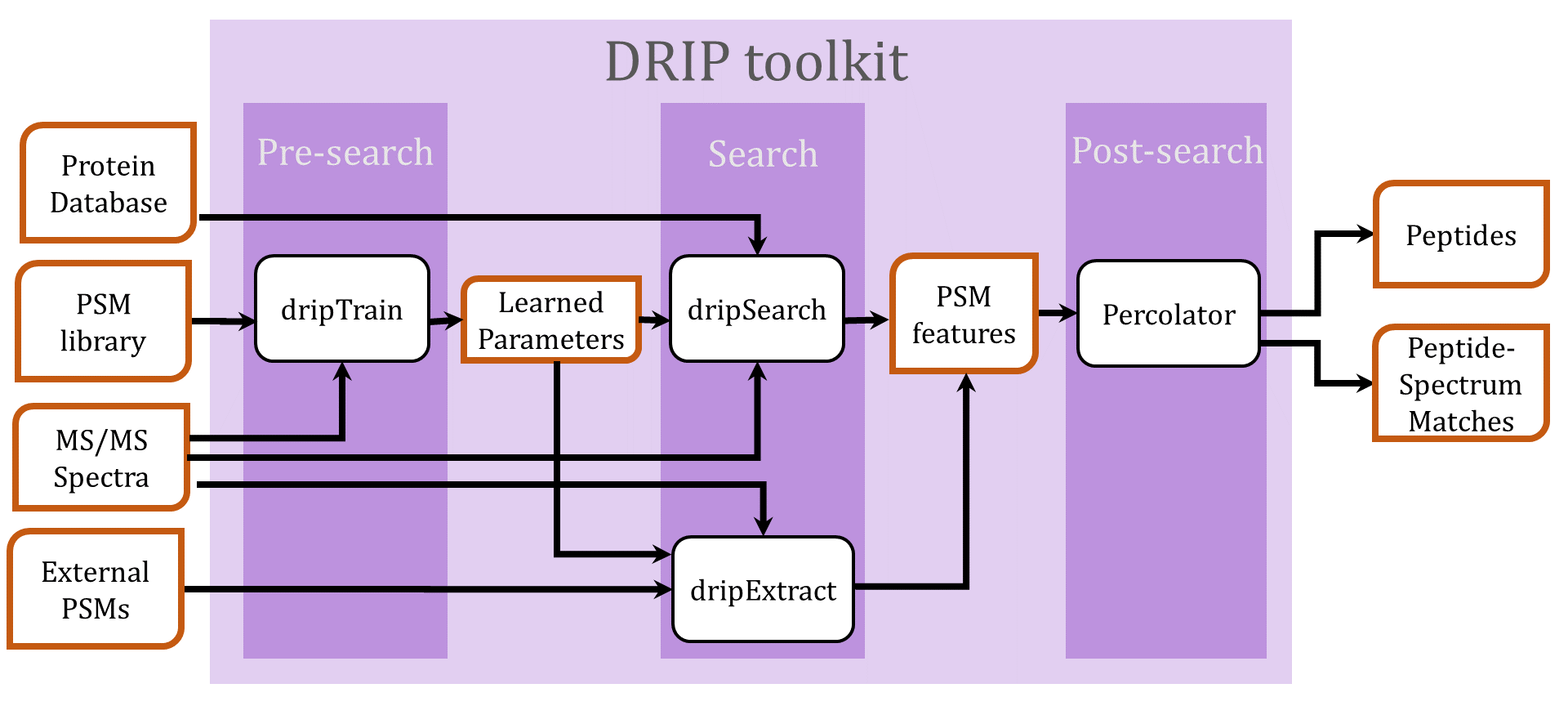
The DRIP Toolkit utilizes a dynamic Bayesian network (DBN) for Rapid Identification of Peptides (DRIP) in tandem mass spectra. Given an observed spectrum, DRIP scores a peptide by aligning the peptide's theoretical spectrum and the observed spectrum, i.e., computing the most probable sequence of insertions (spurious observed peaks) and deletions (missing theoretical peaks). DBN inference is efficiently performed utilizing the Graphical Models Toolkit (GMTK), which allows easy alteration of the model. If you use the DRIP toolkit in your research, please cite:
John T. Halloran, Jeff A. Bilmes, and William S. Noble. "Learning Peptide-Spectrum Alignment Models for Tandem Mass Spectrometry". Thirtieth Conference on Uncertainty in Artificial Intelligence (UAI 2014). AUAI, Quebic City, Quebec Canada, July 2014.Download the DRIP Toolkit View the DRIP Toolkit source here. The toolkit is provided as a set of python modules modules:
| Primary modules | |
| dripDigest.py |
Create an index of all peptides in a fasta file, for use in subsequent
calls to dripSearch.py. |
| dripTrain.py | Train DRIP model parameters given a set of high-confidence PSMs. Only applicable for low-resolution MS2 spectra. |
| dripSearch.py |
Search a collection of spectra against a sequence database, provided
as the output of dripDigest.py, returning a collection of
peptide-spectrum matches (PSMs). |
| dripExtract.py | For input collection of PSMs, derive DRIP features for Percolator analysis. |
| dtk.py | Library for plotting decoded DRIP PSMs, interactive analysis via the Lorikeet plugin, and instantation/manipulation/real-time decoding of DRIP PSMs in the python interactive shell. |
File formats
| dripDigest | dripTrain | dripSearch | |
| MS2 | in | in | |
| Tab-delimited training PSM file (sample) | in | ||
| GMTK learned parameters | out | in | |
| FASTA (sample) | in | ||
| database index | out | in | |
| Tab-delimited text (sample) | out |
DRIP Toolkit documentation
Please send comments and questions to halloj3@uw.edu.